The technology of
sperm cryopreservation, despite the wide variety of cryoprotectors, is not
perfect. The cryopreservation process leads to a loss of approximately 50% of
sperm viability [1]. The role of sperm cryopreservation is associated with
maintaining the initial motility of sperm and preserving metabolism. However,
during the process of cell cryopreservation, damage to proteins, lipids and
nucleic acids is observed [2, 3, 4]. Oxidative processes increase during
cryopreservation. Free radicals damage cellular and subcellular structures,
which reduces cell viability [5]. Oxidative processes are triggers of
morphological and biochemical cryogenic damage, cause sperm dysfunction and
determine the need for the use of antioxidants. Antioxidants limit the effect
of the oxidation process. The use of molecular hydrogen (H2) as a
universal antioxidant is widely discussed today, and its therapeutic effect
based on antioxidant properties is considered in various fields of medicine [6,
7]. However, the rationale for the effectiveness of using H2 in
cryopreservation has not yet been presented in modern literature. An objective
assessment of the effect of H2 is possible when conducting a
comprehensive analysis of the state of spermatozoa. Interference methods of
optical microscopy allow studying cellular processes [8]. Visualization of
structural and morphological changes in cells with high spatial resolution is
an informative way to study the physiological and biomechanical properties of
cells [9]. Interference laser microscopy and interference images make it
possible to evaluate transparent bioobjects with an analysis of the density of
intracellular structures [10].
The aim of the work
was to study the morpho-functional state of spermatozoa during cryopreservation
under the action of H2 using laser interference microscopy.
Interference
microscopy analyzes the morphology of an object depending on the refractive
index of the intracellular structures of cells. The result of interference
microscopy is the registration of the optical density of an object by a direct
and reflected beam of light. This is a fundamental difference from conventional
light microscopy. Conglomerates and dense structures determine an increase in
the refractive index and the registration of convex domains. Oblique
illumination from different angles is used for registration (Fig. 1). The phase
reconstruction is carried out using the phase step method [11]. In this case,
the photodetector registers the displacement of the studied object beam
relative to the reference beam with a known phase, which represents the
interferogram of the object.
The calculation of
the spatial distribution of the intensity of the resulting object in the plane
is carried out according to the following formula:
|
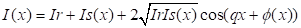
|
(1)
|
Where Ir and Is are
the intensities in the reference and object arms of the interferometer, q is
the spatial frequency of the fringes, φ is the phase associated with
the object.

Fig. 1. Automated interference microscope. 1 – automated two-coordinate stage; 2 – support mirror on piezoelectric element; 3.5 – CCD cameras; 4 – microinterferometer; 6 – laser illuminator.
For transparent
objects, Is(x) has a weak dependence on x. By adjusting the magnification of the
system, it is possible to select a frequency q close to or greater than the
maximum frequency of the interference fringes, limited by the numerical
aperture of the objective, so that the fundamental diffraction resolution of
the resolving power is preserved. The interference term can be separated by
high-pass Fourier filtering. It follows that the complex analytical signal
associated with the real function u(x) can be obtained as
|
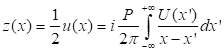
|
(2)
|
In equality (2),
using the Hilbert transform for u(x), the phase associated with the complex
analytical signal z can be represented as follows:
|
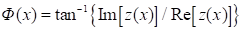
|
(3)
|
The basic basis of
the interference microscope is a modified Mach-Zehnder interferometer. The
radiation source in the Mach-Zehnder interferometer is a He-Ne laser [8]. The
reference field for creating the interference image is tilted relative to the
object beam at an angle of 45° relative to the x and y axes (Fig. 2).
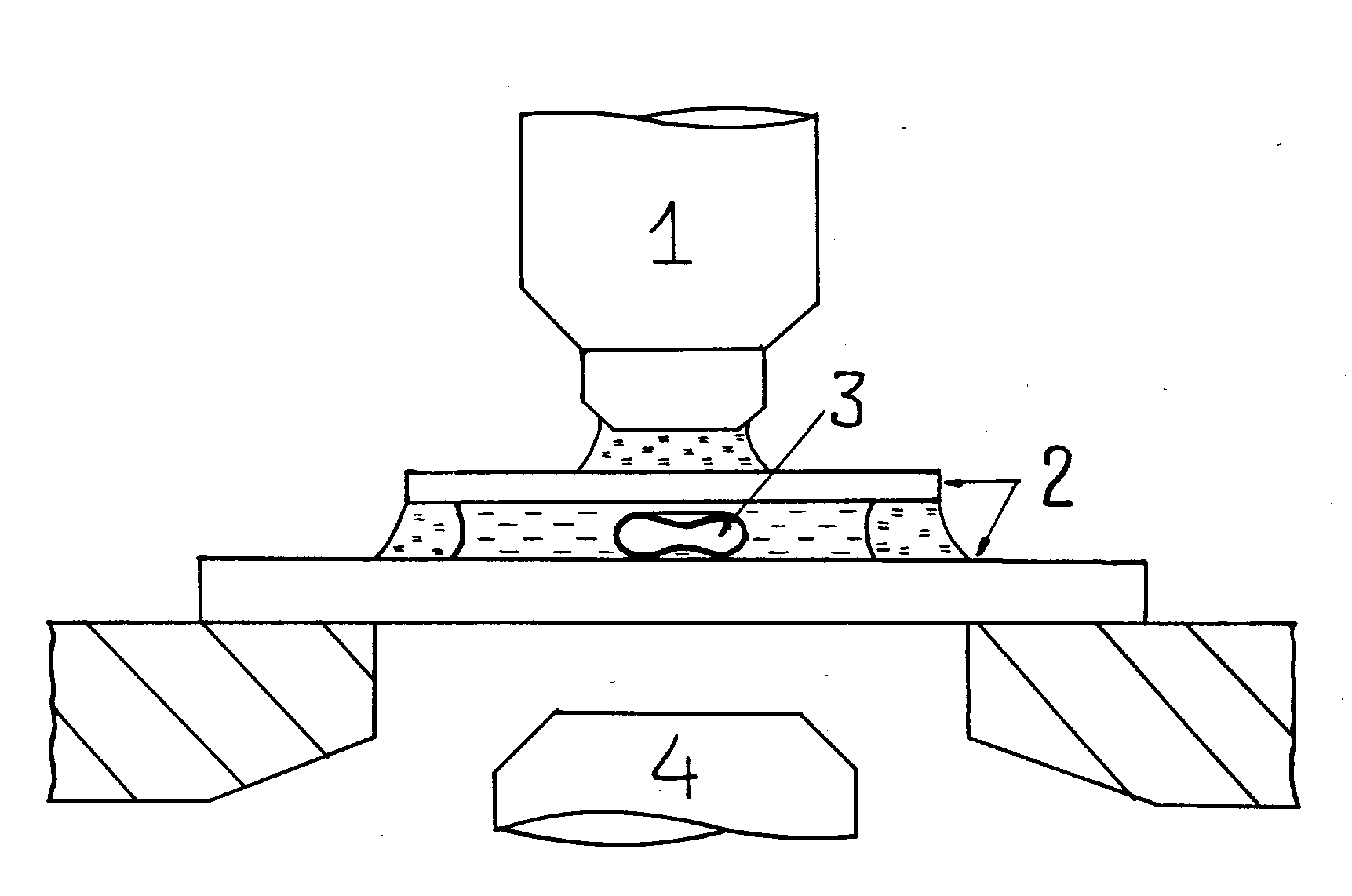
Fig. 2. Schematic representation of the microchamber and (on an enlarged scale) the cell being photometered. 1 – microscope objective, 2 – cover slips, 3 – erythrocyte, 4 – condenser.
The diagram of the
laser interference microscope used in this work is shown in Fig. 3. The device
includes a light source I, semitransparent mirrors BS1, BS2, BS3, rotating
mirrors M1, M2, polarizers WP1, WP2, phase modulator PM, objective O, analyzer
A and detector D. Polarizers WP1, WP2 modulate the phases of the reference and
object beams. Phase modulator PM forms the phase modulation of the reference
beam. The object beam is reflected from the object of study located on the
stage S, passes through mirror BS3, mixes with the reference beam and passes
through analyzer A, which selects from it a component with one or another
polarization, and hits the detector.
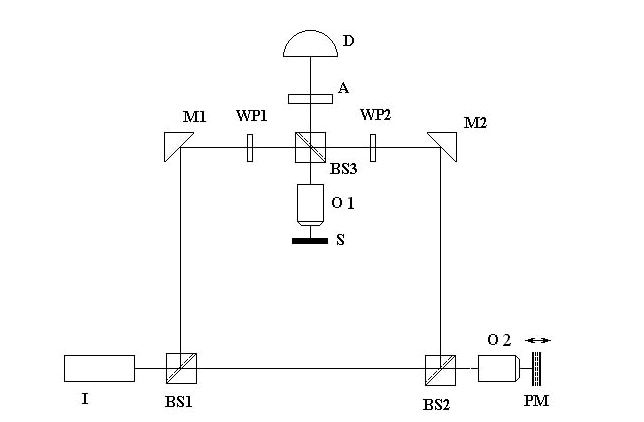
Fig. 3. Schematic diagram of the laser
interference microscope interferometer.
The use of an
interference laser microscope allows for ultra-high resolution, which reaches
0.1 nm (vertically) and 15 - 100 nm in the plane of the object [12]. Calculation
of the interference pattern obtained from the object model allows to represent
the expression of the three-dimensional shape of the cells. This method is a
new approach to visualization for the analysis of cell morphology and can be
useful for studying their unique physiological and biomechanical properties.
The study involved
sperm diluted with the BioXcell medium (France) and then frozen in liquid
nitrogen (-196°C) according to GOST 26030-2015. The condition of the
spermatozoa was analyzed after defrosting the sperm using standard technology.
The study involved sperm diluted with the BioXcell diluent (group I), sperm
after deep freezing (group II), and sperm after deep freezing pre-treated with H2 (group III). The concentration of H2
in the solution was within 1.2-1.5 mg/l. The hydrogen generator «Sputnik-3» (China) produced H2.
To assess the
qualitative parameters of spermatozoa, we used the SFA-500 and Biola AFS-500
sperm analyzers from NPF BIOLA (Russia). The energy parameters of spermatozoa
were assessed by the concentration of ATP using a non-enzymatic method [13].
The antioxidant system was analyzed by the activity of SOD [14] and catalase
[15]. The oxidative properties of cells were determined by the concentration of
MDA in spermatozoa using a reaction with thiobarbituric acid [16].
Interference laser
microscopy was performed on a MIM-340 microscope using a laser with a
wavelength of 650 nm. The vertical resolution was 0.1 nm. Interferograms were
processed in the MIM Visualizer 1.0 program (MIM Software Inc., USA) [17].
Differences between
groups were compared using the Student's t-test with Bonferroni correction,
taking into account the significance threshold of p≤0.05.
Correlation analysis was performed using
the Spearman correlation coefficient.
The calculated
parameters of sperm interforegrams without treatment (group I) were: phase
height 24.03±0.02 nm, capitulum and tail length of spermatozoa 9.53±0.62
μm and 46.82±5.25
μm, respectively (Table 1). Cryopreservation
determined a decrease in the capitulum and tail length in 7.01% and 9.53% of
spermatozoa, respectively (p≤0.05). The use of H2 in the
cryopreservation process led to the preservation of the optical and geometric
parameters of spermatozoa at the level of native cells (group I).
Table 1 The effect of cryopreservation and molecular hydrogen
on the optical-geometric parameters of spermatozoa
|
Sperm
Parameters
|
Native
spermatozoa (group I)
|
Spermatozoa after cryopreservation (group
II)
|
Spermatozoa after exposure to molecular
hydrogen and cryopreservation (group III)
|
|
Capitulum
length, µm
|
9,53±0,62
|
8,32±0,55*
|
9,04 ±0,49
∆
|
|
Tail length, µm
|
46,82±2,05
|
42,36±2,35*
|
45,74± 1,59
∆
|
|
Phase
height, nm
|
24,03±0,02
|
21,32±0,05*
|
23,04 ±0,04*,
∆
|
Note:* – differences in
relation to group I, p≤0.05; ∆ – differences between groups after
cryopreservation (group II and group III).
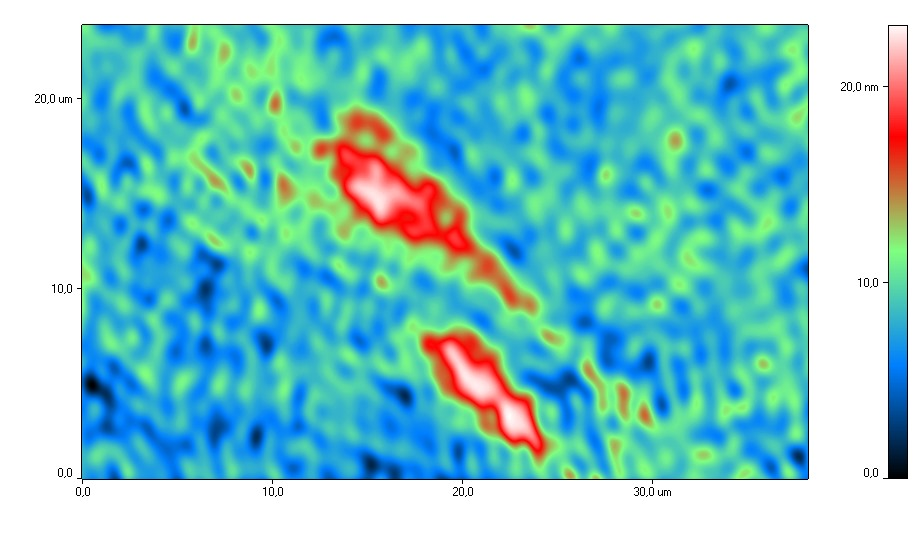
Figure 4 shows
typical phase portraits of spermatozoa exposed to H2
during
cryopreservation and without H2
during cryopreservation. Analysis of
phase interference images of spermatozoa showed that after H2
exposure the capitulum was oval-shaped, the middle part of the cell was thin
and the tail was straight. The acrosomal region after exposure to molecular
hydrogen was well defined and occupied from 40 to 70% of the cell. This
distribution of the acrosome corresponds to the physiological norm, since the
acrosome was shown to cover about 2/3 of the anterior surface of the head.
Determination of the acrosome status in cryopreserved sperm is of fundamental
importance, since cryopreservation directly damages the sperm membranes which
can lead to the loss of the contents of the acrosomal matrix.
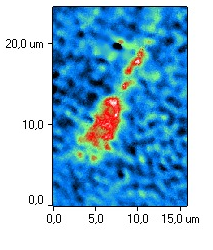
After cryopreservation without H2,
uneven distribution of cytoplasm in the capitulum with abnormal acrosome was
noted. This indicates a change in the permeability of the plasma membrane and
loss of the ability to attach to the oocyte membrane. The loss of acrosomal
matrix content reduces the longevity of cryopreserved spermatozoa [18].
|
|
|
|
a)
|
b)
|
Fig. 4. Typical phase images of
spermatozoa after cryopreservation using H2
(a) and after
cryopreservation without H2
(b).
Evaluation of phase
heights of sperm interphoregrams and changes in the refractive index allowed us
to estimate the distribution of the substance density in the cells. The phase
height in the plane represents the spatial modulation of the wave from a
coherent source. Different refractive indices of parts of the cell are
transformed into a two-dimensional distribution of the optical component [19].
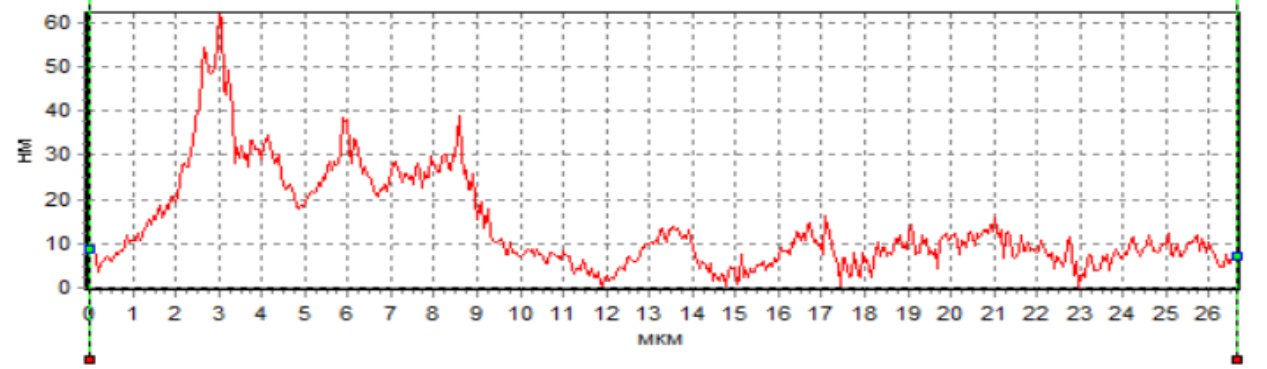
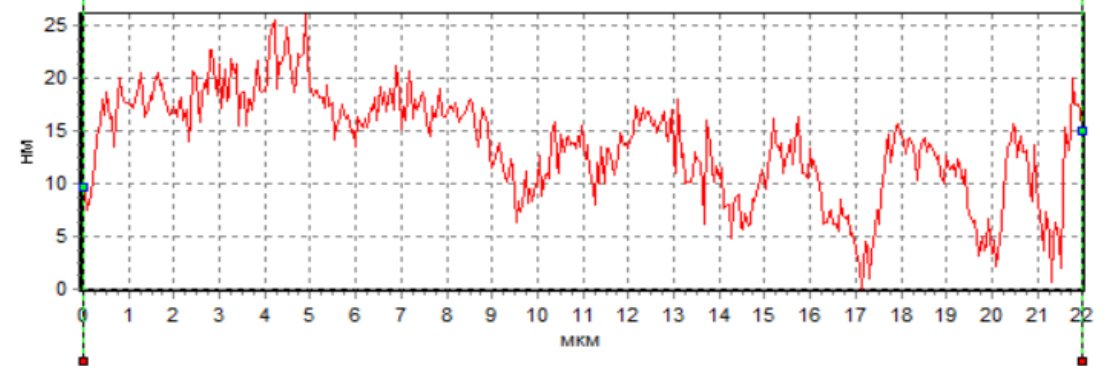
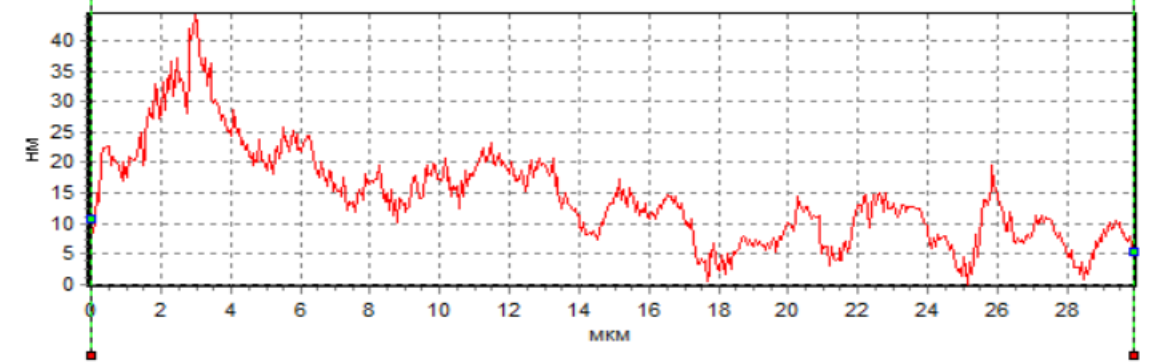
The analysis of the
sperm profile showed the presence of a phase peak in the region of the initial
head segment in native spermatozoa and under the action of H2 during
cryopreservation (Fig. 5a, 5c). This indicates the maximum density in the
acrosomal region. The phase height in the acrosome region of native spermatozoa
and during cryopreservation with H2 significantly exceeded the phase
height in spermatozoa during cryopreservation without H2 (Fig. 5).
This region corresponds to the nucleus with the nucleolus. However, the
decrease in the phase height of spermatozoa during cryopreservation without H2
indicates a decrease in the density in this region of the cell. The low density
is probably due to protein loss. Proteins are involved in fertilization
processes: motility, acrosome reaction, fusion with the egg [20]. Changes in
the protein composition of the sperm membrane are one of the main reasons for
the decrease in sperm fertility after cryopreservation.
Fig. 5. Profiles of changes in the phase
height of spermatozoa: intact (a), after cryopreservation without H2
(b) and after cryopreservation using H2
(c).
This position is
confirmed by previously identified facts obtained using interference
microscopy, indicating that the change in phase height is proportional to the
product of the membrane thickness and the difference in the local refractive
index of the cell and the solution [21].
In our experiments,
a decrease in the phase height of spermatozoa was observed during
cryopreservation (Fig. 5b). Apparently, this is due to damage to spermatozoa
during cryopreservation and their subsequent thawing. In particular, it was
noted that phase heights decrease proportionally to the degree of damage and an
increase in the amount of water inside the cell, which leads to a decrease in
the refractive index due to the loss of concentration of the substance inside
[22]. Destruction of the plasma membrane during cryopreservation can cause
further damage to cells and, consequently, lead to irreversible damage to their
integrity. In many ways, this development of events can be due to oxidative
processes and changes in cell metabolism, since interforegrams reflect the
dynamics of various intracellular processes [23]. To substantiate the stated
position, we conducted biochemical studies of intracellular metabolism in
parallel with the interference registration of cells.
A
study of the oxidative metabolism level revealed that cryopreservation was
accompanied by a significant increase in the lipid peroxidation process, which
was characterized by an increase in the concentration of malondialdehyde (MDA)
by 39% relative to native spermatozoa (group I) (Table 2). The use of H2
in the cryopreservation process made it possible to maintain the intensity of
oxidative processes at the level of native spermatozoa.
Table 2. Oxidative and metabolic activity of spermatozoa
during cryopreservation and exposure to molecular hydrogen
|
Indicators
|
Native
spermatozoa (group I)
|
Spermatozoa after cryopreservation (group II)
|
Spermatozoa after exposure to molecular
hydrogen and cryopreservation (group III)
|
|
MDA,
nmol/ml
|
0,61±0,12
|
0,85±0,14*
|
0,56±0,08 ∆
|
|
SOD, units/mg protein
|
0,61±0,08
|
0,78±0,08*
|
0,87±0,08*, ∆
|
|
Catalase,
mcat/mg
|
9,03±0,76
|
8,48±0,82
|
15,78±0,71*, ∆
|
|
ATP, μmol/l
|
0,79±0,09
|
0,28 ±0,05*
|
0,47±0,04*,
∆
|
Note:* – differences in relation to group
I, p≤0.05; ∆ – differences between groups after cryopreservation
(group II and group III).
The inhibiting
factor of oxidative processes is the increase in the activity of antioxidant
enzymes. The activity of SOD and catalase in spermatozoa increased after the
addition of H2
to the cryopreservation medium (group III) (Table 3).
The activity of SOD and catalase increased by 42% and 74%, respectively
(p≤0.05) relative to native spermatozoa (group I).
The ATP
concentration in spermatozoa after cryopreservation was lower than in native
spermatozoa (group I) by 65%. The ATP content in spermatozoa increased by 67%
during cryopreservation with H2 (group III) relative to group II.
A correlation
dependence was revealed between the metabolic indices and the phase
characteristics of spermatozoa during cryopreservation and the use of H2
against the background of cryopreservation (Table 3). Analysis of the
correlation dependences between the phase height and the oxidative metabolism
index revealed a close negative correlation in all groups (R=-0.88 – Group I,
R=-0.85 – Group II, R=-0.93 – Group III) and a strong correlation between the
phase height and the energy metabolism index (R=0.88 – Group I, R=0.85 – Group
II, R=0.93 – Group III).
Table 3 Correlation analysis of phase height and metabolic
indices in different groups
|
|
Phase Height/MDA
|
Phase Height/ATP
|
|
Native spermatozoa (group I)
|
-0,88
|
0,84
|
|
Spermatozoa after cryopreservation (group II)
|
-0,85
|
0,86
|
|
Spermatozoa after exposure to molecular hydrogen and cryopreservation (group III)
|
-0,93
|
0,91
|
Correlation
relationships prove that the analysis of the phase height of spermatozoa
obtained by interference microscopy allows us to evaluate the total metabolic
activity of spermatozoa and has the highest sensitivity compared to biochemical
methods.
When analyzing the
results obtained, it should be taken into account that during cryopreservation,
lipids and proteins in a liquid state harden, turning into a gel, forming a
rigid and fragile structure that is more sensitive to damage [24].
Our study shows that during
cryopreservation, there is a loss of integrity of both acrosomal and plasma
membranes. During freezing, spermatozoa undergo significant metabolic changes
and mitochondrial function is greatly impaired [25]. The bioenergetic function
of mitochondria plays an important role in spermatozoa, especially for
capitation, hyperactivation and acrosome reaction [26]. The obtained results
demonstrate that the use of H2
is effective in protecting sperm
metabolism during cryopreservation. The use of H2
can ensure the
preservation of sperm functional parameters.
The study showed
that the use of H2
during cryopreservation maintained the number of
motile spermatozoa, the number of fast spermatozoa, and the average speed of
spermatozoa at the level of native cells (Table 4). Whereas after
cryopreservation of spermatozoa, these indicators were reduced.
Table 4. Effect of molecular hydrogen on sperm fertility
parameters
|
Sperm
Fertility Criteria
|
Native
spermatozoa (group I)
|
Spermatozoa after cryopreservation (group
II)
|
Spermatozoa after exposure to molecular
hydrogen and cryopreservation (group III)
|
|
|
|
|
Mobility, %
|
82,51±3,95
|
71,15±3,34*
|
79,62
±
3,60
∆
|
|
|
Number of mobile, million/dose
|
35,76±2,17
|
27,35±2,16*
|
33,71±2,03
∆
|
|
|
Number of fast, million/dose
|
65,54±7,14
|
51,77±6,13*
|
58,98±6,55
∆
|
|
|
Average
speed, µm/sec
|
85,62±3,54
|
74,53±2,48*
|
81,56±3,52
∆
|
|
Note:* – differences in
relation to group I, p≤0.05; ∆ – differences between groups after
cryopreservation (group II and group III).
Considering that the
speed of sperm movement is one of the most informative indicators of sperm
quality [27], the detected increase in the number of motile, fast spermatozoa
and the average speed of spermatozoa under the influence of H2 compared to
these indicators during cryopreservation proves the effectiveness of using H2
as a cryoprotector.
1. The study
demonstrated the effectiveness of using H2
as a new strategy for
protecting spermatozoa during cryopreservation.
2. Analysis of
spermatozoa interforegrams provides a comprehensive assessment of the state of
spermatozoa metabolic processes during cryopreservation.
3. Phase images
allow for clear identification of spermatozoa with a reduced functional state
which can be used for express analysis of spermatozoa quality.
This work was
supported by the Russian Science Foundation (project No. 23-26-00205).
1. Watson P.F. The causes of reduced fertility with cryopreserved semen // Anim Reprod Sci. 2000. № 60–61. Р. 481–92. doi: 10.1016/s0378-4320(00)00099-3.
2. Sion B., Janny L., Boucher D., Grizard G. Annexin V binding to plasma membrane predicts the quality of human cryopreserved spermatozoa // Int J Androl. 2004. №27 (2). Р. 108–1014. doi: 10.1046/j.1365-2605.2003.00457.x.
3. Kogan T., Dahan D.G., Laor R., Argov-Argaman N., Komsky-Elbaz A., Kalo D., Roth Z. Association between Fatty Acid Composition, Cryotolerance and Fertility Competence of Progressively Motile Bovine Spermatozoa // Animals (Basel). 2021. №11 (10). Р. 2948. doi: 10.3390/ani11102948.
4. Thomson L.K., Fleming S.D., Aitken R.J., De Iuliis G.N., Zieschang J.A., Clark A.M. Cryopreservation-induced human sperm DNA damage is predominantly mediated by oxidative stress rather than apoptosis // Hum Reprod. 2009. № 24(9). Р. 2061–70. doi: 10.1093/humrep/dep214
5. Partyka A., Lukaszewicz E., Nizanski W., Twardon J. Detection of lipid peroxidation in frozen-thawed avian spermatozoa using C(11)-BODIPY(581/591) // Theriogenology. 2011. №75 (9). Р. 1623–9. doi: 10.1016/j.theriogenology.2011.01.002.
6. Zhai X., Chen X., Shi J., Shi D., Ye Z., Liu W. Lactulose ameliorates cerebral ischemia-reperfusion injury in rats by inducing hydrogen by activating Nrf2 expression // Free Radic. Biol. Med. 2013. № 65. Р. 731–741. doi: 10.1016/j.freeradbiomed.2013.08.004
7. Miller M. W., Sadeh N. Traumatic stress, oxidative stress and post-traumatic stress disorder: neurodegeneration and the accelerated-aging hypothesis // Mol. Psychiatry 2014. № 19. Р. 1156–1162. doi: 10.1038/mp.2014.111
8. Lue N., Popescu G., Ikeda T., Dasari R., Badizadegan K., Feld M. Live cell refractometry using microfluidic devices // Opt. Lett. 2006. № 31(18). Р. 2759-2761. doi: 10.1364/ol.31.002759
9. Park Y., Popescu G., Badizadegan K., Dasari R., Feld M. Diffraction phase and fluorescence microscopy // Opt. Expr. 2006. № 14(18). Р. 8263-8268. doi: 10.1364/oe.14.008263
10. Levin G.G., Bulygin F.V., Vishnyakov G.N. Coherent oscillations of the state of protein molecules in living cells // Tsitology. 2005. V.47. №4. P.348-356.
11. Deryugina A.V., Ivaschenko M.N., Ignatiev P.S., Metelin V.B., Talamanova M.N. Possibilities of intravital visualization of blood cells under stress to assess the state of the body // Scientific visualization.2023. V. 15. № 1. P. 90-99. doi: 10.26583/sv.15.1.08
12. Tychinsky V.P., Kretushev A.V., Klemyashov I.V., Vyshenskaya T.V., Shtil A.A., Zatsepina O.V. Coherent phase microscopy – a new approach to studying the physiological state of the nucleolus // DAN. 2005. № 405(4). P. 432-436.
13. Deryugina A.V., Danilova D.A., Pichugin V.V., Brichkin Yu.D. The effect of molecular hydrogen on functional states of erythrocytes in rats with simulated chronic heart failure // Life. 2023. №3(13). Р. 418. doi: 10.3390/life13020418
14. Sirota T.V. Standardization and regulation of the rate of superoxide-generating reaction of adrenaline autoxidation used to determine the pro/antioxidant properties of various materials // Biomedical Chemistry. 2016. № 6. P. 650–655. doi: 10.18097/PBMC20166206650
15. Deryugina A.V., Abaeva O.P., Romanov S.V., Vedunova M.V., Ryabova E.N., Vasenin S.A., Titova N.A. Electrokinetic, oxidative and aggregation properties of red blood cells in the postoperative period following kidney transplantation // Russian journal of transplantology and artificial organs. 2020. № 2. P. 72-79. doi: 10.15825/1995-1191-2020-2-72-79
16. Deryugina A.V., Efimova T.S., Boyarinov G.A., Nikolskiy V.O., Kuznetsov A.B., Simutis I.S. Correction of metabolic indicators of erythrocytes and myocardium structure with ozonized red blood-cell mass // Cell and Tissue Biology. 2018. V. 12. № 3. P. 207-212. doi: 10.1134/S1990519X18030033
17. Deryugina A.V., Belov A.A., Ivashchenko M.N., Ignatiev P.S., Metelin V.B. Assessing the functional state of red blood cells by using the laser interference microscopy // Cell and Tissue Biology. 2021. V. 15. № 4. P. 388-392. doi: 10.1134/S1990519X21040027
18. Bailey J.L., Bilodeau J.F., Cormier N. Semen cryopreservation in domestic animals: a damaging and capacitating phenomenon // J. Androl. 2000. № 21. Р. 1-7.
19. 19. Tychinsky V. P. Dynamic phase microscopy: is a “dialogue” with a cell possible? // Uspekhi Fizicheskikh Nauk. 2007. №5 (177). P. 535–552 doi: 10.3367/UFNr.0177.200705c.0535
20. Kondoh G., Tojo H., Nakatani Y., Komazawa N., Murata C., Yamagata K., Maeda Y., Kinoshita T., Okabe M., Taguchi R. Takeda J. Angiotensin-Converting Enzyme Is a GPI-Anchored Protein Releasing Factor Crucial for Fertilization // Nature Medicine. 2005. № 11. Р. 160-166. https://doi.org/10.1038/nm1179.
21. Yusipovich A.I., Berestovskaya Yu.Yu., Shutova V.V., Levin G.G., Gerasimenko L.M., Maksimov G.V., Rubin A.B. New possibilities of studying microbiological objects by laser interference microscopy // Biophysics. 2011. V. 56. № 6. P. 1091–1098.
22. Zagubizhenko M.V., Yusipovich A.I., Pirutin S.K., Minaev V.L., Kudryashov Yu.B. Using the method of laser interference microscopy to study the state of mouse peritoneal macrophages irradiated with ultraviolet light // Radiation biology. Radioecology. 2011. № 6(51). P. 715–720
23. Brazhe A.R., Brazhe N.A., Sosnovtseva O.V., Pavlov A.N., Mozekilde E., Maksimov G.V. Study of cellular dynamics using interference microscopy with wavelet analysis // Computer research and modeling. 2009. V. 1. № 1. P. 77–83
24. Gao D., Critser J.K. Mechanisms of cryoinjury in living cells // ILAR J. 2000. №41(4). Р. 187–96. doi: 10.1093/ilar.41.4.187.
25. Schober D., Aurich C., Nohl H., Gille L. Influence of cryopreservation on mitochondrial functions in equine spermatozoa // Theriogenology. 2007. № 68. Р. 745–54. doi: 10.1093/ilar.41.4.187.
26. O’Flaherty C., de Lamirande E., Gagnon C.. Positive role of reactive oxygen species in mammalian sperm capacitation: triggering and modulation of phosphorylation events // Free Radic Biol Med. 2006. № 41(4). Р. 528–40. doi: 10.1016/j.freeradbiomed.2006.04.027
27. Lyashenko A.A. Biological parameters of bull sperm depending on the storage period in liquid nitrogen // Zootechnical science of Belarus. 2015. V. 50. №1. С. 126-134.